КАТАЛИТИЧЕСКИХ РЕАКЦИЙ КИНЕТИКА. Каталитич. реакция - циклич. процесс, складывающийся из ряда элементарных реакций, скорости которых описываются действующих масс законом. Этот закон имеет простую форму для идеальных газовых смесей, идеальных растворов и идеальных поверхностных слоев. Последним термином обозначают модель (И. Ленгмюр, 1918), в которой пов-сть твердого тела принимается состоящей из определенного числа одинаковых мест (адсорбц. центров), каждое из которых способно удержать в адсорбир. состоянии одну частицу - молекулу или атом; при этом частицы, адсорбир. на соседних местах, не влияют друг на друга. Если в элементарной реакции номер s участвуют вещества
А1 и А2 из объемной фазы (газовой или жидкой), вещества I1 и I2 из поверхностного слоя, занимающие на пов-сти доли и соотв., то скорость реакции
где ks - константа скорости, [A1] и [А2] - концентрации А1 и А2; a1, а2, i1, i2 - числа молекул веществ, участвующих в реакции; q0-доля своб. пов-сти (или, при реакции в растворе, доля пов-сти, занятой растворителем); z - число своб. мест пов-сти, которые требуются в дополнение к (i1+i2) местам, занятым молекулами I1 и I2, для образования активир. комплекса, т.е. переходного состояния (при гетерог. реакции активир. комплекс подобен адсорбир. молекуле, однако может занимать на пов-сти более одного места). Фактически в реакции не участвует более 3 частиц, так что те или иные сомножители в (1) всегда отсутствуют. Если отсутствуют , и q0, кинетич. ур-ние (1) определяет скорость гомог. реакции, если эти сомножители присутствуют, - скорость гетерог. реакции. Ур-ние для rs, содержащее только величины q, иногда наз.
законом действующих поверхностей. Ур-ние (1) описывает также процессы адсорбции и десорбции: если адсорбируется вещество A, z = 1 и rs = ks [A]q0; если А десорбируется, z = 0 и r-s=k-sqА (индексом — s обозначают величины, относящиеся к элементарной реакции, обратной по отношению к реакции номер s).
В случае неидеальных систем правая часть (1) содержит дополнительно т. наз. кинетич. фактор активности, равный произведению коэф. активности вступающих в реакцию веществ, деленному на коэф. активности активир. комплекса.
Зависимость от температуры констант скорости элементарных реакций описывается Аррениуса уравнением; предэкспоненц. фактор этого ур-ния м. б. вычислен или приближенно оценен как для гомогенных, так и для гетерогенно-каталитич. реакций с помощью активированного комплекса теории. Скорость каталитич. реакций в идеальной системе вычисляют на основе найденных по ур-нию (1) скоростей составляющих ее элементарных реакций (см. Сложные реакции). В пром. реакторах каталитич. реакции почти всегда стационарны или квазистационарны (см. Квазистационарности приближение). Скорость гетерогенно-каталитич. реакции относят к единице площади пов-сти катализатора. Для техн. целей ее рассчитывают на единицу массы катализатора или на единицу объема слоя зерен катализатора (см. Активность катализатора). Часто итог каталитич. реакции выражается двумя или более хим. ур-ниями. В таких случаях для расчета скорости образования целевого продукта важна селективность катализатора.
В данной статье К. р. к. рассматривается в предположении, что реакция происходит в кинетич. области, т. е. на ее скорость не влияют процессы переноса вещества и тепла. Учет этих процессов, существенно влияющих на скорость мн. гетерогенно-каталитич. реакций, рассматривается в ст. Макрокинетика.
Сопоставление кинетич. ур-ний с опытными данными выполняют по-разному в зависимости от типа реактора. При использовании загрузочного реактора периодич. действия, являющегося замкнутой системой, кинетич. ур-ние интегрируют по времени, обычно используя приближение квазистационарности. В проточном трубообразном реакторе осуществляется стационарная реакция; кинетич. ур-ние интегрируют по объему слоя катализатора, обычно в приближении режима идеального вытеснения, т. е. предполагая, что скорость движения объемной фазы одинакова по всему сечению реактора и можно пренебречь продольной диффузией. Надобность в интегрировании и в указанных упрощающих предположениях отпадает при использовании безградиентных реакторов, позволяющих измерять непосредственно скорость реакции при данных концентрациях реагентов и продуктов.
Гомогенный катализ. Гетерогенный катализ на однородных поверхностях. Наиб. просты каталитич. реакции, состоящие из двух стадий:
Здесь Z в случае гомог. катализа - молекулакатализатора, в случае гетерог. катализа - место на пов-сти катализатора (адсорбц. центр), A1 и А2 - исходные вещества, B1 и В2 - продукты реакции, I - промежут. частица. Стрелками обозначены элементарные реакции; в итоговом ур-нии ставится знак равенства. Скорости элементарных реакций выражаются равенствами:
r1=kl[Z][A1]; r-1=k-1[ZI][B1];
r2 = k2[ZI][A2];r-2 = k-2[Z][B2] (2)
Кроме того, имеется уравнение баланса:
[Z] + [ZI] = [Z]S, (3)
где [Z]S в случае гомог. катализа - суммарная (определяемая аналит. методами) концентрациякатализатора. В случае
гетерог. катализа [ZI] и [Z] в (2) обозначают q1 и q0 и ур-нию (3) соответствует равенство: q1+q0=1, так что [Z]S=1. При этом пропорциональность скорости реакции числу мест на пов-сти учтена в значениях констант k1, k-1, и т. д. Для реакций с участием газов принято вместо [A1], [А2] вводить в ур-ния типа (2) парциальные давления, . Если принять, что реакция, протекающая по схеме (1), стационарна, то для ее скорости можно записать:
r = r1-r-l= r2-r-2. (4)
Ур-ния (2)-(4) дают:
В частных случаях в схеме (1) B1 или А2 (либо оба эти вещества) могут отсутствовать. Если отсутствует B1 (при этом I совпадает с А1) принимают B1=1 и т. п. Возможно также, что вместо А1 будут два вещества - А'1 и A''1; тогда полагают [A1]=[A'1][А''1] и т. п. Так, схема (1) отражает простейший механизм ферментативного катализа:
где Е-фермент, S-субстрат, Р-продукт. Предполагая реакцию в замкнутой системе квазистационарной и применяя равенство (5), получаем ур-ние Михаэлиса (1913):
где kM = (k-1+k2)/k{-T. наз. константа Михаэлиса. Ур-ние (7) описывает кинетику мн. ферментативных реакций. Пример гетерогенно-каталитич. реакции с простым двустадийным механизмом - конверсия СО водяным паром на среднетемпературном (ок. 400 °С) катализаторе, гл. составной частью которого является Fe3O4:
В (8) Z обозначает кислородную вакансию на пов-сти Fe3O4, которая образуется, когда молекула СО, ударяясь о пов-сть, удаляет с нее атом О (стадия 2). Вакансия вновь заполняется атомом О из Н2О (стадия 1). В схеме (8) стадии пронумерованы так, чтобы она согласовалась с механизмом (1); при стационарной реакции нумерация стадий произвольна, т. к. все элементарные реакции происходят одновременно. Кислородные вакансии составляют лишь малую долю пов-сти, т. е. q0 >> q0; тогда из (2) и (4) следует:
Предполагая поверхностный слой идеальным, получаем из (5):
При равновесии (r= 0)
где К -константа равновесия. Отсюда ур-ние (9) можно представить в след, форме:
где k = k1k2/k-2 и а=kl/k-2.
Если при стационарно протекающей по схеме (1) реакции изменить скачком концентрации веществ в объемной фазе, а затем поддерживать их постоянными, то ZI и Z будут
постепенно приближаться к значениям, соответствующим новому стационарному состоянию; при этом скорость реакции будет изменяться по ур-нию:
где t-время от момента изменения условии реакции, r: - новая стационарная скорость реакции (достигаемая формально при t=:), r0-скорость реакции немедленно после изменения ее условий, t - постоянная, наз. временем релаксации скорости реакции:
В случае гетерог. катализа вместо [Z]S надо ввести в (13) L - число мест на единице площади пов-сти (-); при этом скорости элементарных реакций должны быть определены как число актов реакции на единице площади пов-сти за единицу времени.
На основе (13) можно показать, что t [ 25u, где u=[Z]S/r: (при гомог. катализе) или u=L/r: (при гетерог. катализе); u наз. временем оборота катализатора. При этом предполагается, что изменение скорости реакции вызвано лишь изменением концентрации промежут. веществ, т. е. релаксация скорости реакции является "собственной". При гетерогенно-каталитич. реакциях возможны также "сторонние" процессы релаксации скорости реакции, вызванные обратимым изменением катализатора из-за изменения объемной фазы, которое приводит к изменению констант скорости элементарных реакций. Напр., при окисленииэтилена в этилен-оксид кислородом на серебре сторонний релаксац. процесс обусловлен обратимым поглощением кислородасеребром. Он значительно медленнее собственного релаксац. процесса.
В случае реакций с механизмом более сложным, чем выражаемый схемой (1), в отсутствие сторонних релаксац. процессов, как правило, т не превышает и. Известны также автоколебат. реакции, у которых стационарная скорость вовсе не устанавливается. В идеальных системах это возможно лишь при условии, что механизм реакции содержит стадию взаимод. разл. промежут. веществ друг с другом (считая промежут. веществами также своб. места пов-сти); тогда становится возможным существование более одного стационарного состояния (см. Колебательные реакции).
Катализ на неоднородных поверхностях. Модель идеального адсорбир. слоя, как наиболее простая, широко применяется для описания кинетики гетерогенно-каталитич. реакций. Она, однако, не всегда способна описать количественно явления на пов-стях реальных катализаторов. Напр., скорость адсорбции не пропорциональна q0, а зависит от нее экспоненциально. В таких случаях необходимо отказаться по крайней мере от одного из предположений модели идеального адсорбир. слоя: либо считать места пов-сти неодинаковыми (т. наз. биографич. неоднородность), либо принять возможность взаимного влияния адсорбир. частиц (индуцир. неоднородность пов-сти). Тогда термин "однородная поверхность" будет означать почти то же, что и "идеальный адсорбир. слой", однако допускается возможность адсорбции частицы на двух и более соседних местах пов-сти.
Согласно модели биографически неоднородной пов-сти, равновесие адсорбции вещества I на однородной пов-сти (обозначается Z+I=ZI) подчиняется закону действующих масс:
(q1/q0P1)=Ka (14)
Т. к. q0+q1=1, ур-ние (14) дает гиперболич. изотерму адсорбции Ленгмюра (см. Адсорбция). Константу адсорбц. равновесия Ка наз. адсорбц. коэффициентом. Для места биографически неоднородной пов-сти роль q1 играет вероятность а того, что это место занято частицей I. Неоднородность пов-сти м. б. описана с помощью величины x=-lnКа, наз. показателем десорбируемости; чем 4 больше, тем молекула I легче десорбируется. Произведение
j(x)dx, где j(x) - дифференц. ф-ция распределения мест пов-сти по показателю десорбируемости, есть число мест пов-сти единичной площади, для которых показатель десорбируемости вещества I принимает значения в пределах от до x + dx. Пусть x0 - наименьшее, x1 - наиб. значение x на данной пов-сти, так что если x < x0 или x > x1, то j(x)=0. Для описания опытных фактов достаточны ф-ции распределения в интервале x0 < x < x1 двух видов:
j(x)=A (15)
(равномерная неоднородность),
j(x) =Aеgx, 0<g<1 (16)
(экспоненциальная неоднородность), где А - постоянная, g=T/q, q - постоянная, имеющая размерность температуры Т. Распределение (16) формально переходит в (15) при g=0. Можно также допустить, что -1<g<1. Для области средних покрытий, для которьгх при x=x0 s@1, при x=x1 s@0, распределение (15) приводит к логарифмич. изотерме адсорбции Темкина, а распределение (16) - к степенной изотерме Фрейндлиха. Гиперболич. изотерма Ленгмюра обычно не описывает адсорбц. равновесие на катализаторах.
Связь константы равновесия К элементарной реакции Z+A:ZI+B с ее константой скорости k определяется соотношением:
k=gKa, 0<a<1, (17)
где g и a - постоянные, a не зависит от Т. Постоянную a часто наз. коэф. переноса, а (17)-правилом переноса. Соотношение, аналогичное (17), связывает также k с Ка, т. к. К пропорциональна Ка. Единичный акт элементарной реакции сопровождается уменьшением на единицу числа своб. мест пов-сти. Для обратной реакции, при которой место освобождается, коэф. переноса обозначают р. Из условий хим. равновесия следует, что для каждой стадии s as+bs=1, gs=g-s Зависимости, аналогичные (17), установлены экспериментально Й. Брёнстедом для реакций гомог. кислотно-основного катализа, М. Поляни и Н. Н. Семеновым - для серий сходных гомог. реакций с участием атомов и своб. радикалов, А. Н. Фрумкиным - для электродных процессов; теоретич. интерпретация этих зависимостей дана М. Эвансом и М. Поляни.
Чтобы получить кинетику реакции (1) на неоднородной пов-сти, для упрощения принимают, что коэф. переноса стадий 1 и 2 одинаковы: a1=a2=a. Интегрирование вкладов разл. мест пов-сти в скорость реакции дает:
где m=a-g (0<m<1), f=x1-x0 - т наз. показатель неоднородности пов-сти, k01, k02, k0-1, k0-2 - значения соотв. констант скорости при x=0. Если g=0 (равномерная неоднородность), то g/(еgf-1) заменяется на 1/f, а m - на a. В области малых покрытий (у всех мест s<<1) скорость реакции, протекающей по схеме (1), описывается ур-нием (18) с m=0; для области больших покрытий, в которой у всех мест s@1, - ур-нием (18) с m=1. В обоих случаях выражение для r по форме не отличается от получаемого из (5), различны лишь значения постоянных. Этим объясняется то, что ур-ние (9) согласуется с опытными данными, хотя, как показывает исследование равновесия стадии 1 схемы (8), пов-сть катализатора при снятии кислорода равномерно неоднородна. В качестве примера реакции на неоднородной пов-сти в области средних покрытий рассмотрим синтез NH3. Если парциальное давление NH3 не слишком мало, адсорбцияазота является стадией, определяющей скорость процесса, и механизм реакции м. б. представлен схемой:
равновесная стадия 2 в действительности состоит из стадий гидрирования адсорбир. азота, но это не существенно для вывода кинетич. ур-ния, т. к. равновесие не зависит от механизма, по которому устанавливается. Р-ция происходит при средних покрытиях пов-сти азотом и малых покрытиях водородом и промежут. продуктами гидрирования, поэтому применимо ур-ние (18). Квазиравновесность стадии 2 означает, что k0-2>>k01PA1; k02PA2>>k0-1PB1. T. к. PA1=PN2, PA2=P3H2, РВ1=1, РB2=P2NH3, получаем:
или
где k+ и k- - постоянные. При равновесии r=0, следовательно, k+/k-=К, где К - константа равновесия реакции N2 + ЗН2 = 2NH3. Она не зависит от характеристик катализатора, поэтому ур-ние (20) содержит лишь 2 постоянные, т и k+ (или k-), подлежащие определению из результатов измерений скорости реакции. Для промотированных железных кат., применяемых в промышленности, обычно получают m = 0,5; при этом a=0,5 и g=0. Ур-ние (20) подтверждено опытами при давлениях от 0,025 до 50 МПа. При более высоких давлениях требуется ввести поправку, учитывающую отклонение газов от идеальности и влияние давления на константы скорости. Правило переноса (18) выполняется для данной элементарной реакции не только при переходе от одного места неоднородной пов-сти к другому, но также (хотя и более грубо) при переходе от одного катализатора к другому. Если к стадиям реакции, происходящей по схеме (1) на разл. однородных пов-стях, применимо правило переноса (18), скорость реакции максимальна на таком катализаторе, для которого . Следовательно, при необратимости стадий и при g1=g2, a1=a2=0,5 и PА1@PA2 скорость реакции максимальна, если стандартное изменение энергии Гиббса в первой стадии DG01 примерно равно половине стандартного изменения энергии Гиббса для реакции в целом. Сходное утверждение, но относящееся к тепловым эффектам стадий и реакции в целом, наз. принципом энергетич. соответствия (впервые сформулирован А. А. Баландиным).
Лит.. Уолтер Ч., Кинетика ферментативных реакций, пер. с англ., М., 1969; Снаговский Ю. С., Островский Г М., Моделирование кинетики гетерогенных каталитических процессов, М., 1976; Киперман С Л., Основы химической кинетики в гетерогенном катализе, М., 1979; Яблонский Г С., Быков В. И., Горбань А. Н., Кинетические модели каталитических реакций, Новосиб., 1983; Тёмкин М И., "Кинетика и катализ", 1984, т. 25. в. 2,с. 299 305; Temkin М. I., "Advances in Catalysis", 1979, v. 28, р. 173 281; Boudart M., Djegamariadassou G.. Cinetique des reactions en catalyse heterogene, P., 1982. М.И. Тёмкин.
 и
и 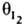 соотв., то скорость реакции
соотв., то скорость реакции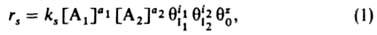
 ,
,  и q0, кинетич. ур-ние (1) определяет скорость гомог. реакции, если эти сомножители присутствуют, - скорость гетерог. реакции. Ур-ние для rs, содержащее только величины q, иногда наз.
и q0, кинетич. ур-ние (1) определяет скорость гомог. реакции, если эти сомножители присутствуют, - скорость гетерог. реакции. Ур-ние для rs, содержащее только величины q, иногда наз.
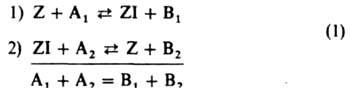
 ,
,  . Если принять, что реакция, протекающая по схеме (1), стационарна, то для ее скорости можно записать:
. Если принять, что реакция, протекающая по схеме (1), стационарна, то для ее скорости можно записать:
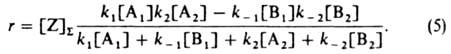





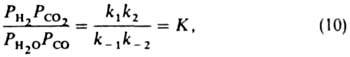

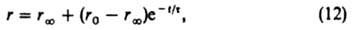
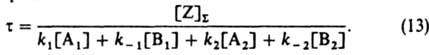


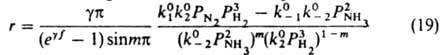

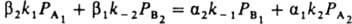 . Следовательно, при необратимости стадий и при g1=g2, a1=a2=0,5 и PА1@PA2 скорость реакции максимальна, если стандартное изменение
. Следовательно, при необратимости стадий и при g1=g2, a1=a2=0,5 и PА1@PA2 скорость реакции максимальна, если стандартное изменение